The advent of Magnetic Resonance Imaging (MRI) revolutionized the way medical practitioners diagnose and track diseases throughout the body. MRI utilizes magnetic properties of ions in the body along with computer-generated radio waves to create detailed images of the body’s organs and tissues4. This allows for the detection of cancers, traumatic brain injury, strokes, aneurysms, spinal cord disorders, and other ailments, without exposing patients to radiation or necessitating the use of intravenous dyes as required in other forms of diagnostic imaging. While many advances in MRI technology have been made to implement artificial intelligence for image reconstruction, increasing magnetic field strengths, optimizing receiver coil arrays, and enhancing imaging gradients, there remains an ongoing need to prioritize expanding access of these technologies on a global scale.
One area of advancement in MRI research that has received recent attention is the use of lower field-strength (0.2 Tesla) MRI systems2,3. These systems were once thought to provide suboptimal imaging quality as they utilize a substantially lower magnetic field strength compared to modern MRI systems. Integration of artificial intelligence for low-field MRI systems provides the capability for its images to compete with the resolution of that of a high-field MRI2. There are several advantages to low-field MRI that directly impact healthcare facilities and the patients they serve. Importantly, low-field MRI does not require a cooling system nor a large energy source in order to function properly1,2,3,5. This allows for a reduction in the ongoing costs associated with MRI systems in addition to a reduction in the high maintenance fees (~$10 thousand per month) and acquisition costs (~$1million/T) that are required of high-field MRI systems1,3,5. For under-resourced healthcare centers, these fees can be the determining factor for whether or not a patient receives a lifesaving diagnostic scan.

The utility of low-field MRI systems extends beyond cost savings, however. Due to the smaller magnetic field, noise produced by these systems is reduced which favors its use among pediatric populations1,3,5. In 2020, the FDA approved the use of the first portable point-of care low-field MRI (Below is a video of Dr.Kevin Sheth, a critical care neurologist at Yale School of Medicine discussing the advent of a the world’s first portable low-field MRI). Its small footprint and open design allows for family members to remain at the bedside with patients as they receive their scan1,2,3,5. The small footprint of these systems also makes its use in preclinical research settings more accessible. The open design of these systems is an additional benefit for patients with claustrophobia as well as obese patients that have difficulty in high-field MRI systems. Widescale clinical use of low-field MRI would expand access for patients that have metal implants such as pacemakers or shunts, who otherwise would not receive such diagnostic imaging2. Given the ability of a portable low-field MRI system to provide cost savings to healthcare facilities, expand access to patients in need, and further diagnostic capabilities for practitioners, low-field MRI systems are posed to pioneer a new era of medical diagnostic imaging.
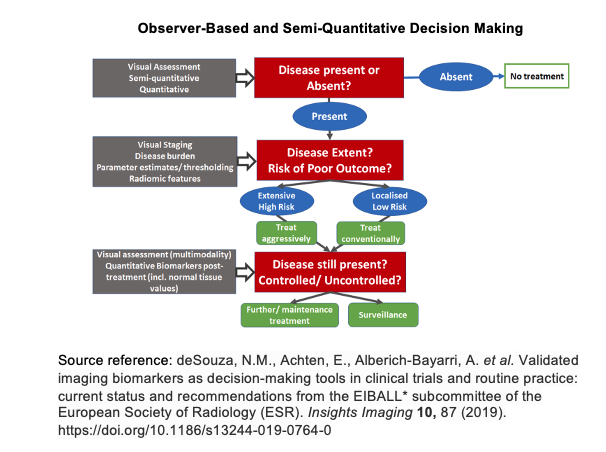
Incorporating artificial intelligence into low-field MRI diagnostic imaging stratifies the detection of disease by combining the observer-based image interpretation currently in practice with an artificial intelligence generated semi-quantitative approach (please see observer-based and semi-quantitiative decision making diagram below). In doing so, as larger datasets of diagnostic images are collected, artificial intelligence algorithms become more reliable in detecting disease pathology. Such measures may be used to not only detect disease but to better inform clinicians of potential treatment responses and health outcomes of their patients.
- Cooley CZ, McDaniel PC, Stockmann JP, Srinivas SA, Cauley SF, Śliwiak M, Sappo CR, Vaughn CF, Guerin B, Rosen MS, Lev MH, Wald LL. A portable scanner for magnetic resonance imaging of the brain. Nat Biomed Eng. 2020 Nov 23. doi: 10.1038/s41551-020-00641-5. Epub ahead of print. PMID: 33230306.
- Ghadimi M, Sapra A. Magnetic Resonance Imaging Contraindications. [Updated 2020 May 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK551669/
- Grist, T. M. (2019). The Next Chapter in MRI: Back to the Future? Radiology, 293(2), 394-395. doi:10.1148/radiol.2019192011
- J.P. Hornak, The Basics of MRI, Interactive Learning Software, Henrietta, NY, 2020, http://www.cis.rit.edu/htbooks/mri/.
- Sarracanie, M., & Salameh, N. (2020). Low-Field MRI: How Low Can We Go? A Fresh View on an Old Debate. Frontiers in Physics, 8. doi:10.3389/fphy.2020.00172
- Sheth KN, Mazurek MH, Yuen MM, et al. Assessment of Brain Injury Using Portable, Low-Field Magnetic Resonance Imaging at the Bedside of Critically Ill Patients. JAMA Neurol. Published online September 08, 2020. doi:10.1001/jamaneurol.2020.3263