
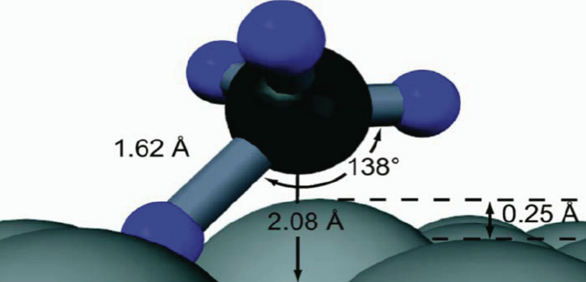
Our experiments are designed to answer a variety of important questions regarding metal catalysts:
- What is the geometric structure of the transition state?
- How do atoms move during reaction?
- What is the molecular-level mechanism for reaction?
- Is the surface a static template or an active participant?
- How quickly can energy flow within the reaction complex?
- Can we exploit energy flow to control reactivity?
- How accurate are theoretical predictions of reactivity?
Our Chambers
We use ultra-high vacuum (UHV) chambers equipped with powerful surface science instrumentation to help us elucidate these molecular-level details. Though the two chambers in our lab are similar, each chamber is outfitted with unique equipment allowing us to work on multiple projects at once.
Chamber 1
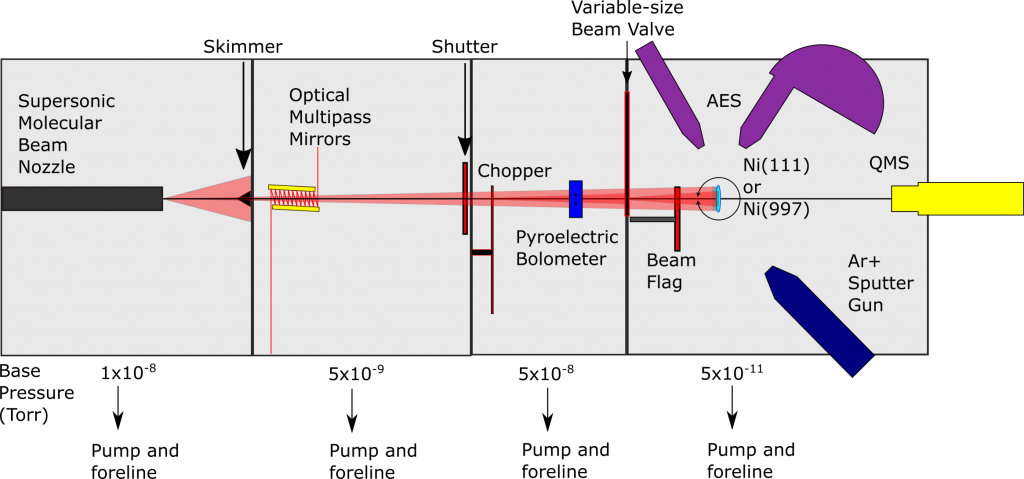
Initially designed with the idea of state-resolved molecular beam experiments, Chamber 1 sought to understand the transition state geometry and vibrational efficacy towards the reactivity of methane on nickel surfaces. Establishing that vibrations have a profound impact on reactivity, the precise control over the impinging methane molecules also allowed for subtle surface temperature effects to appear. Studies have largely focused on the Ni(111) single-crystal whose atomically flat surface makes for a simplified system of study while also being the most common surface site on industrial nanoparticles. Though the (111) surface facet is of great interest, it neglects the quantity of defect sites present in the nickel nanoparticles. By using a lightly-stepped Ni(997) surface, the effects of steps and terraces on methane reactivity may be elucidated.
Steam reforming also contains a nontrivial percentage of other hydrocarbons besides methane. Due to the complexity of the degrees of freedom present in larger hydrocarbons, the reactivity of C2 species such as ethane is of interest, and the most recent studies on Chamber 1 have focused on this topic. Ultimately, these experiments provide insight to the experimental limits of state-resolved chemical reactivity studies while also furthering our understanding of industrially significant reaction of activated C-H bond cleavage.
Chamber 2
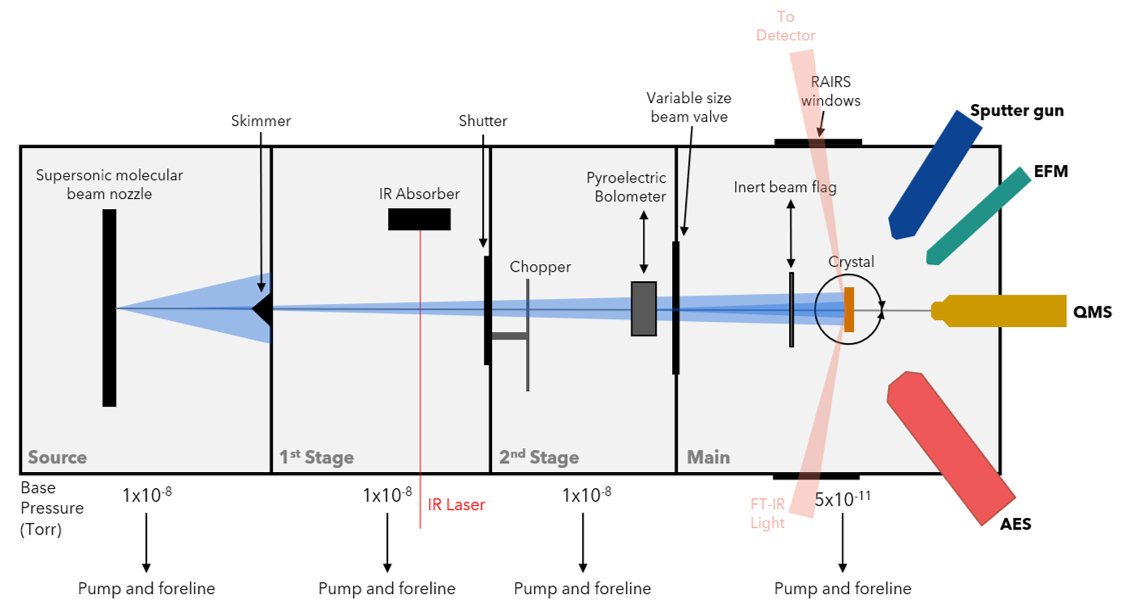
Chamber 2 has a nearly identical capabilities to Chamber 1, but in 2019 the Main stage was been modified to enable the study of C-H bond cleavage on single-atom alloys (SAAs). A flux-monitored evaporator in combination with an FT-IR spectrometer for reflection/absorption spectroscopy (RAIRS) permits gas-surface scattering experiments on SAAs allowing us to collect site-specific reactivity information. Our extensive studies of methane on single-crystal surfaces have provided a foundation for extending our knowledge of the reactivity of hydrocarbons and small molecules on SAA surfaces.
Chamber 2 was historically used to study the effects of thermally excited vibrations in methane on Ni(111) and Ir(111) single-crystal surfaces as well as trapping-mediated dissociation on Ir(110). The group then switched to a Cu single-crystal to study small molecule and alkane reactivity on RhCu SAAs.
Main focuses:
–
Bond- and Mode-Selective Chemistry
Bond Selectivity
By pre-exciting the C-H stretch and measuring the product yield for CHD3 dissociation on Ni(111), we find the yield of C-H bond cleavage products to be nearly 100-fold greater than C-D bond cleavage products. Exciting the C-H stretch in the molecule dictates product identity.
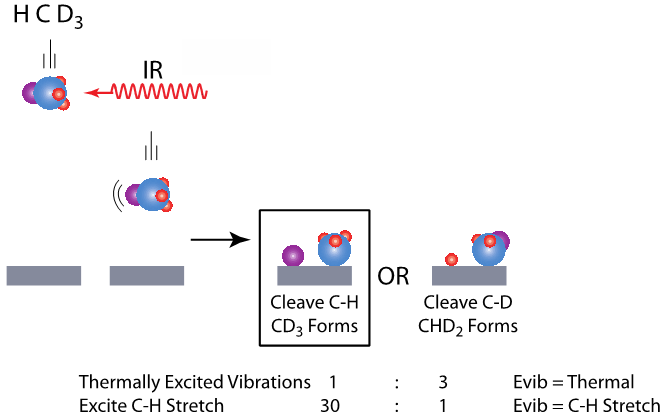
Vibrational Mode Selectivity
Vibrational modes differ in their ability to promote reaction, such as in CH4 where v3 (C-H stretch) promotes C-H bond cleavage much more than 3v4 (bend) which indicates that a molecule’s vibrational state influences the reactivity (sticking probability, S0). This is called mode selectivity. For mode selective chemistry, the initial Evib must persist long enough to enhance TS access and the vibration must enhance access to distorted TS geometry. Both bond- and mode-selective chemistry occur because there is too little time during the molecule-surface encounter (ca. 200 fs or less) for Evib to be redistributed.
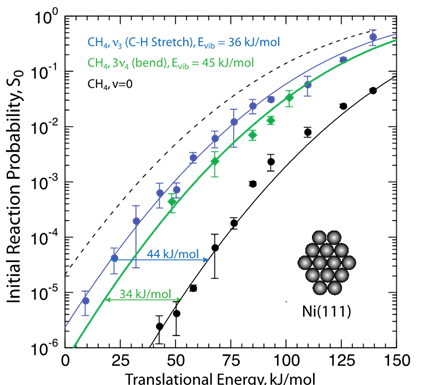
–
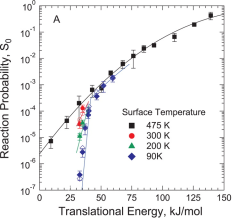
Surface Temperature Effects
Commercial steam reforming reactors operate at temperatures around 1000K, and the first C-H bond cleavage in methane dissociation on the Ni catalyst is believed to be the rate-limiting step. Despite the commercial importance of this reaction, nearly all studies probing the dynamics of methane dissociation have focused on surface temperatures of 600K or lower. We have studied the impact of surface temperature on methane activation over a broad range of surface temperatures including those approaching industrial levels (82 K ≤ TS ≤ 1000 K). Our use of methane molecules with precisely defined energy provides a clear view of how surface temperature, and therefore surface atom motion, impacts reactivity.
Carbon Dissolution
A distinctive characteristic of nickel is the ability for carbon to diffuse into the subsurface and bulk layers of the crystal. In methane steam reforming, the build-up of carbon in the subsurface of the nickel catalyst leads to a gradual reduction in surface reactivity and ultimately deactivation of the catalyst itself. Additionally, the initial dissolution and subsequent reemergence of carbon from the subsurface are key steps in the growth of well-ordered graphene on nickel substrates via chemical vapor deposition (CVD). While researchers have previously used AES and XPS to study the dynamics of carbon dissolution on nickel surfaces, we employ beam reflectivity measurements to monitor the process in real time.
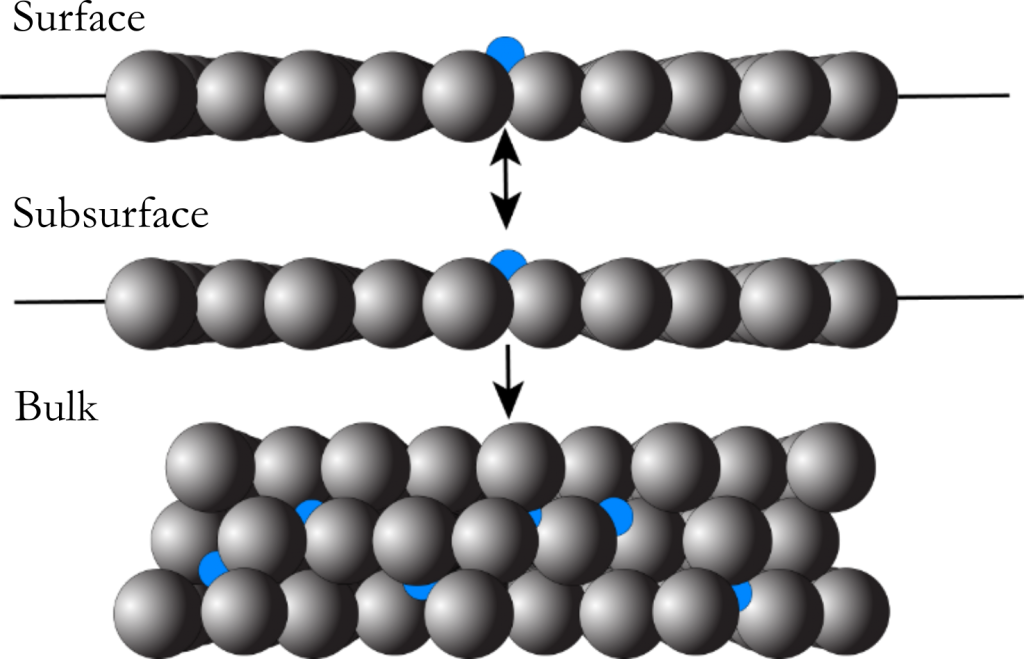
We observe significant changes in the reaction profile by increasing surface temperature as the rate of dissolution approaches the reactive flux of the high energy gas molecules. We use these results to further develop kinetic models for methane reactivity as a function of surface coverage as well as carbon diffusion into the nickel crystal. The major parameters from these models include the site-blocking coverage and its subsequent dependence on surface temperature as well as an updated measure of the barrier to diffusion for the C/Ni system.
– –
Reaction Energetics
Studies exploring how vibrational energy Evib promotes chemical reactivity most often focus on molecular reagents, leaving the role of substrate atom motion in heterogeneous interfacial chemistry underexplored. Using a combination of theoretical and experimental studies on methane dissociation in Ni(111), it can be shown that lattice atom motion modulates the reaction barrier height during each surface atom’s vibrational period, which leads to a strong variation of the reaction probability S0 with surface temperature. The results show the first direct measurement in a beam-surface scattering experiment that reveal a sharp energetic threshold that permits a direct comparison with density functional theory (DFT) calculations for barrier height.
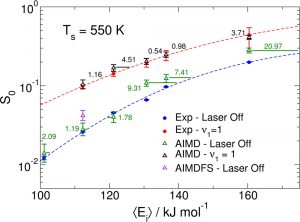
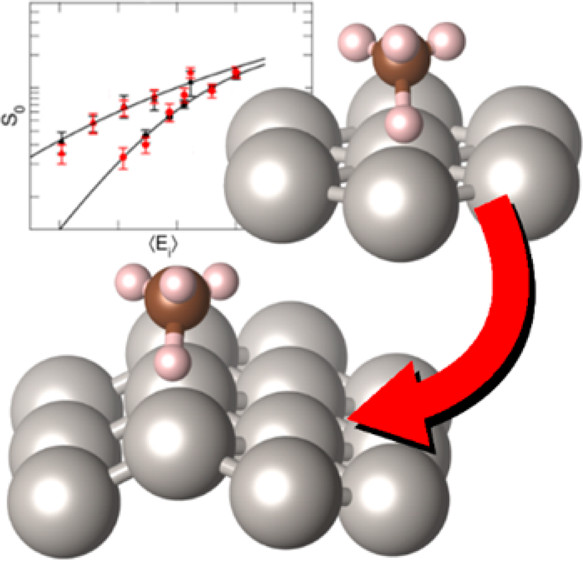
Accurately simulating heterogeneously catalyzed reactions requires reliable barriers for molecules reacting at defect sites, such as steps. Unfortunately, theory has struggled to compute accurate barriers in this case. Predictions of reaction rates and dynamics are contingent on the accuracy of potential energy surface projections. However, dissociative chemisorption of polyatomic reagents on metal is more complicated, relying on the electronic structure of the metal, van der Waals interactions, lattice motion effects, and the participation of numerous degrees of freedom.
By combining state-resolved molecular beam experiments with theory, specifically the ab initio molecular dynamics (AIMD) using specific reaction parameter density functional theory (SRP-DFT), reliable molecule-metal interactions can be determined. The experimental and theoretical initial state-selected sticking measurements are within 4 kJ/mol of each other. The accuracy on a flat (111) surface can also be extended to stepped (211) surfaces to emulate defects, which bridges a gap between fundamental surface science studies and real-life heterogeneous catalysts that often proceed over defected metal nanoparticles.
–
Single-Atom Alloy Catalysis
Individual atoms of precious metals dispersed on a more inert metal host form a single-atom alloy (SAA) surface that exhibits unique catalytic selectivity and activity when compared to their monometallic counterparts. SAAs are a new generation of heterogeneous catalysts that show promising potential to catalytic insights due not only to their increased activity (and in some cases, selectivity) but also cost- and resource-effectiveness. Key mechanistic details of gas-surface interactions on SAAs, which would reveal insights into molecular mechanics and energy flow dynamics, remain largely experimentally unexplored. Our lab aims to fill this gap by using state-resolved molecular beam experiments to extract precise information with regard to these atomic-sized kinetic details. We pair our RAIRS-equipped FT-IR spectrometer alongside our supersonic molecular beams to allow for real-time studies of reactions at specific catalytic sites.
Support
This material is based upon work supported by the National Science Foundation under Grant Numbers 1465230 and 1800266. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.