APPENDIX
- Transwell 2D co-cultures are not compatible for confocal/two-photon imaging:
- Glutamate Protocol Table
Arri has reviewed the relevant papers to identify different media compositions that were used. Using this information, we are working on refining the protocol for the glutamate excitotoxicity study. Additionally, given that most of the paper that we found were using 2D cultures, we plan to look at other papers that use 3D cultures to determine if a change of medium is required prior to the injury induction. The upper range of the glutamate concentration we use (300uM) is higher than that mentioned in the papers because, unlike the paper, we will use 3D culture, and 3D diffusion is worse than 2D diffusion.
| Ref | Relevance | Glutamate Concentration | Exposure | Media/rinse | Notes |
| https://www.sciencedirect.com/science/article/pii/S0304394013002152 | HESCs treated with glutamate at physiological concentrations | 200 uM | 24h | DMEM (with supplements) first and then changed to Neurobasal-A (with supplements) 50uM DAPT was included in first medium change | Changed to trophically deprived, glutamate-free mimal medium (90% salt-glucose-glycine medium and 10% MEM) 1 day prior to injury |
| https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6773069/ | NMDA and glutamate on rat cortical culture | 100 and 300 uM | 30 minutes | Control media = MEM + 0.01% BSA + 25mm Hepes + 10 um glycine Rinse: MEM 200:1 dilution | Younger culture is less sensitive so use higher conc. No change of medium prior to experiment After toxicity assay: cells are exposed to control solution, NMDA, or glutamate for 30 mins and then rinse |
| https://www.nature.com/articles/cddis2012194 | rat derived hippocampal, cortical, midbrain neurons | 100 uM glutamate in 10 uM glycine, Mg2+ free medium | 15 minutes | HBSS (hippocampi) Neurobasal medium (Gibco-Invitrogen) supplemented with B-27 (Gibco-Invitrogen) and 2 mM L-glutamine (culture) | No change of medium prior to experiment |
| https://onlinelibrary.wiley.com/doi/full/10.1046/j.1471-4159.2000.0751045.x | mouse cortical cultures | 100 to 500 uM | Didn’t remove, glutamate media because they were testing enzyme degradation | Medium: Neurobasal medium, 2% B27, 0.5 mM L-alanyl-L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin (Life Technologies) + 1% horse serum | No change of medium prior to experiment |
| https://www.nature.com/articles/s41419-018-0351-1 | Rat hippocampal neurons from Wistar rat E18 embryos | 30 and 100 uM (100 is most sig) Also test at 1 and 10 uM but not sig | 30 mins | For low-astrocyte cultures: cytosine with AraC (Sigma-Aldrich) at conc. of 2 uM at DIV2-5 | No change of medium prior to experiment |
| https://journals.biologists.com/jcs/article/131/22/jcs214684/56996/Axonal-degeneration-induced-by-glutamate | (E18 Sprague-Dawley) Rat embryonic hippocampal neurons | 20uM | 6 hours | After 3 h, the plating media was changed to Neurobasal™ medium supplemented with 2% B27, 0.5 mM GlutaMAX™-I and P/S After 3 days, a third of medium was replaced and treated with 5 uM AraC to inhibit glial cell proliferation | No change of medium prior to experiment |
Table 2. Glutamate concentration, media composition, and injury exposure time for glutamate excitotoxicity induction of different cell lines from published literature.
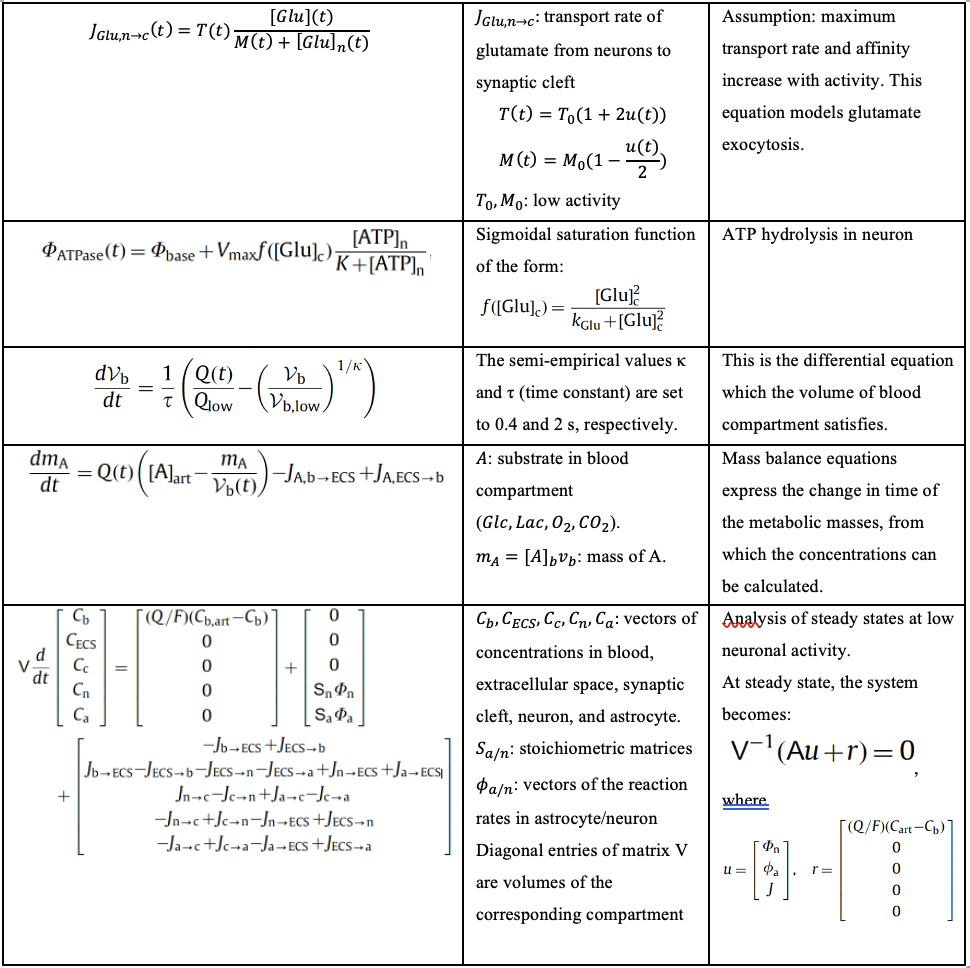
Table 3. List of differential equations involved in TBI which are relevant to our computational model and project.
Sources: (Çakir, 2007) and (Calvetti, 2010)
Cell passaging protocol
Materials needed: DMEM cell culture medium, trypsin, HMC3 cells in cell culture dish.
- Warm the DMEM and trypsin in a 37˚C water bath for 15~20 minutes.
- Spray the biohood workspace w/ 10% bleach, 75% ethanol and wipe surface with tissue paper.
- Applicable to any items brought into the hood
- Aspirate old media from the cell culture dish
- Wash twice with 5 mL PBS
- Aspiration of the PBS should not be in contact with cells attached to the dish bottom
- After adding PPBS, shake disk gently to distribute PBS
- Add 1.5 mL of 0.25% trypsin-EDTA and put the dish in the 37˚C incubator for 2-3 minutes
- After 2-3 minutes, use microscope to check confluency / cell suspension
- Add 8.5 mL of media to the cell culture dish
- Transfer the cells (with trypsin and cell culture media) into a 15 mL conical centrifuge tube
- Centrifuge at 1500 rmp for 3 minutes to pellet the cells
- Aspirate cell culture media in conical centrifuge tube and re-suspend the cell pellet in 5 mL of new cell culture media
- Gently pipette to mix and avoid bubbles
- Calculate the desired seeding ratio and add the corresponding amount of cell suspension and media to the new cell culture dish
- This depends on when the cells will be used
- We plan to independently practice cell passaging in 5 days so we picked a ratio of 1:5 so that sufficient confluency is reached. This means that ⅕ of the cell suspension (0.5mL) and 9.5 mL of new medium are needed for a 10 mL dish
- Extract 0.5 mL of the cell suspension from conical centrifuge tube and gently inject into the new cell culture dish with 9.5 mL of fresh media
- Tilt the dish in north-west-south-east direction to distribute the cells
- This depends on when the cells will be used
- Aspirate and discard the old cell suspension in the centrifuge tube to biohazard bin
- Put the new cell culture dish in the 37˚C incubator overnight
Optical image acquisition protocol
In general, we will measure spectral intensity at 755nm, 860 nm and 910nm excitation, detecting on a multi-wavelength PMT every 10 nm from 490 to 630 nm. We will measure fluorescence lifetime at the same excitations but detect only at 460 and 525 nm using a hybrid detector and a PicoQuant TCPSC module which allows for the high temporal resolution needed for fluorescence lifetime imaging.
- Maintain Excel spreadsheet from centralized code base (on Georgakoudi network drive) to auto-generate file names for control and injury scaffolds.
- Maintain plastic well plate with labeled control and injured scaffold sections and numbered wells to track each ROI throughout timepoints.
- For each scaffold, remove from its place in the well and secure it with a metal harp to a glass bottom dish. Place the smooth (non-cut) side up.
- Set the 40x water objective and add one drop of DI water. Lower the objective z stage fully and secure the dish in the sample stage.
- Search for cells using brightfield eyepiece viewing. Cells are transparent in this mode but the edges can be seen when adjusting the Z height. Once cells are found, turn off the microscope internal light and lower all coverings, then confirm the presence of cells in Live imaging mode
- Set the depth range based on the range of visible cells. Z slices are 4 microns apart.
- Begin with intensity acquisition mode (xyz). All settings (line average, frame average) are set to 1, except for 8 frame accumulation. Speed is 60, bidirectional. The pinhole is at 1 AU (airy units).
- Turn on the transmission PMT and both PMTs and HyDs.
- Set and tune the wavelength for 755 nm.
- Begin acquisition. Save name according to spreadsheet.
- Repeat for 860, changing the depth by +1 micron to adjust for laser co-registration.
- Change to FLIM acquisition mode. Set CFD (voltage) to 40 for 755, 60 for 860. Acquire and ensure that decay traces are smooth with no interruption. FLIM data is saved and renamed individually.
- Change to spectral acquisition mode (xy-lambda). Change detector to the wavelength-adjusting PMT. Open the pinhole to 7.77 AU (full opening).
- After spectral acquisition, change the pinhole back to 1 AU immediately to prevent photobleaching.
- Repeat for as many ROIs are acquired per scaffold.
- Retrieve the plastic well plate from the incubator. Return to the hood. Spray a tweezer with ethanol and dry in the hood. Pick up the scaffold from the edge and return to its original well. Retrieve the next scaffold and continue.
LysoTracker staining for microglia cells protocol
To prepare the HMC3 cell cultures, follow the same protocol for cell passaging, then measure the cells by adding 10 µL of the cell solution to 10 µL trypan blue to measure cell viability in the automatic cell counter. Calculate the volume needed to obtain from the 5 mL cell-medium solution. Add 1 mL of the cells to each well in the well plate with 1 mL medium, then incubate the cells at 37˚C and 5% CO2 overnight.
For the Lysotracker staining, we need to optimize the concentration by the following steps:
- Discard the cell culture medium
- Add 2 mL pre-warmed medium, which contains 10 nM, 20 nM, 50 nM, 75 nM, and 100 nM LysoTracker that was previously mixed in a falcon. This step is to optimize the cell culture concentration.
- Incubate the cells for 30 minutes at 37˚C
- Discard the medium and add 2 mL pre-warmed fresh medium
Then, we proceed to the fixation step:
- Discard the medium and fix the cells by adding 200 µL 4% paraformaldehyde in pre-warmed medium (including FBS) for 15 minutes at 37˚C.
- Discard fixation medium and wash the cells 2 times with 2 mL PBS for 2 minutes each
- Discard PBS and add fresh PBS, then check the cells under the Confocal microscope
After optimizing the concentration, we need to optimize the incubation time. The purpose of optimization is to receive the best signal-to-noise ratio when imaging the cells.
LPS treatment of microglia protocol
Measure the cell number and calculate the volume using the same steps as the LysoTracker staining protocol.
- Add 1 mL of the cells to each well of the 12-well plate and 1 mL medium, then incubate the cells at 37˚C and 5% CO2 overnight
- Add 20 µL LPS at a concentration of 100 µL/mL to each well and incubate for 0h, 24h, or 48h
To generate the LPS solution
- Dissolve 0.1 mg LPS powder in 10 mL sterilized distilled water and vortex for 1 minute to achieve 10,000 ng/mL stock solution (kept at -20˚C in aliquots)
- Based on the final concentration, calculate the volume needed to obtain from the LPS stock in order to stimulate the cells using the following equation
For 50 ng/mL, we need 2 mL * 50 ng/mL = 10,000 ng/mL * x, x = 10 µL
- We need 6 wells for each time point (2 untreated wells, 2 wells treated with 50 ng/µL, and 2 wells treated with 100 ng/µL LPS)
Glutamate treatment of microglia protocol
PREPARATION OF CELL CULTURES FOR IMAGING
Use 1 10cm dish on a 80-90% confluence and split it at 9 wells of each 12-well plate
- Discard the medium from the dish, add 10ml PBS and wash the cells. Discard PBS. Add 1,5 ml trypsin and incubate the cells at 37oC for 3-4 minutes. Check at the microscope that the cells have been detached from the bottom of the dish
- Transfer the cells to a 15 ml tube. Add 8,5 ml medium and centrifuge at 1000 rpm for 5 min. Discard supernatant. Add 3 ml of medium and resuspend the cells.
- Measure the cells by adding 10ul from the cell solution and 10 ul of Trypan Blue and measure cell viability to the automatic cell counter. Amplify the number of cells that you have with 3 (due to total of 3ml medium). For 9 wells you need 9 * 120.000= 1080000 cells in total. So use
In 3ml you have Y total number of cells (=what I see at cell counter*3)
Xab=; 1080000 cells
So you can calculate the volume that you need to obtain from the 3ml in order to have total 1080000 cells. Complete this volume up to 9 number of wells x1ml each well=9ml.
- Then add 1 ml of the cells to each well and add further 1 ml medium to each well and incubate the cells to 37°C and 5% CO2
- Next day add glutamate: 4ul for 10uM glutamate from stock 5mM, 20ul for 100uM glutamate from stock 10mM, 100ul for 500uM glutamate from stock 10mM, 200ul for 1000uM glutamate from 10mM stock.
12-well plate for imaging
A1-A3: 3 wells untreated
B1-B3: 3 wells with X1 concentration glutamate
C1-C3: 3 wells with X1 concentration glutamate
PREPARATION OF CELL CULTURES FOR ROS ASSAY
**Seed cells for ROS ASSAY: USE BLACK 96-well plates with transparent bottom**
From the remaining cells from those used for imaging we need:
3- Xab ul used Total cells Y-1080000
X=? 9×25000=225000
So take the X ul from the tube and complete up to a final volume of 9×200=1800 ul and then add 200 ul per well
FOR 0.5HR (2 wells untreated and 1 well positive control)
A1,2,3 medium
B1,2,3 cells untreated
C1,2,3 cells with 500uM glutamate
D1,2,3 cells with 1000uM glutamate.
E1,2,3 positive control
F1,2,3 medium ONLY no change
FOR 24HR (want 12 wells*25000)
A1,2,3 medium
B1,2,3 cells untreated
C1,2,3 cells with 10uM glutamate
D1,2,3 cells with 100uM glutamate.
E1,2,3 positive control
F1,2,3 medium ONLY no change
FOR 6HR
A1,2,3 medium
B1,2,3 cells untreated
C1,2,3 cells with 100uM glutamate
D1,2,3 cells with 500uM glutamate.
E1,2,3 positive control
F1,2,3 medium ONLY no change
ROS ASSAY
Buffer Preparation :
10X Buffer: Prepare 1X Buffer by diluting 10 mL 10X Buffer with 90 mL ddH2O. Mix gently and thoroughly. Equilibrate to 37°C before use. 1X Buffer can be kept frozen or at 4°C for future use. 1X BUFFER READY FRIDGE
DCFDA Solution. Prepare a working DCFDA solution by diluting 20 mM DCFDA in 1X Buffer: to make a 20 μM final concentration, add 10 μL of 20 mM DCFDA solution to 10 mL 1X Buffer. DCFDA may also be diluted in media without phenol red.
Need 12wells x 100=1200 ul, so better use 2ul of DCFDA at 2ml of 1X buffer. MAKE FRESH – aluminum foil
TBHP Solution (Positive Control). Prepare a 50 – 250 μM TBHP working solution by diluting 55 mM TBHP stock solution in the 1X Supplemented Buffer. Make fresh each time and do not store for future use (storage may lead to TBHP degradation). TBHP may also be diluted in complete media with 10% FBS without phenol red.
50uMx1ml=55000 x; x=0.9 ul in 999 ul of complete media with 10% FBS without phenol red (AB)
** Make all glutamate stocks in complete media with 10% FBS without phenol red **
Remove the media and add 100 μL/well of 1X Buffer.
Remove 1X Buffer and stain cells by adding 100 μL/well of the diluted DCFDA Solution (45 min at 37oC in dark)
Discard DCFDA and add 100 ul of medium at the wells that originally had only medium.
Also add 100 ul of medium at the untreated cells.
Also add 100 ul to each well to the cells with the positive control (100ul from the TBHP that you have generated above – AB).
Make in Eppendorf:
For glutamate 10uM add 1.2 ul from 5mM stock in 598.8ul of medium, add 100 ul per well
For glutamate 100uM add 6 ul from 10mM stock in 594 of medium, add 100 ul per well
For glutamate 500uM add 30 ul from 10mM stock in 570ul of medium, add 100 ul per well
For glutamate 1000uM add 60 ul from 10mM stock in 540ul of medium, add 100 ul per well
Let the dish in the incubator for 0.5 or 6 or 24hours.
Measure right away without discarding the treatment or Discard the medium from wells and add 100 ul of buffer 1x and measure always from bottom.
Same day for 0.5 and 6 hours or next day for 24 hours treatment measure plate immediately on a fluorescence plate reader at Ex/Em = 485/535 nm. Measure from the bottom.