Paper 2
Central Inhibition of IKKβ/NF- κB Signaling Attenuates High-Fat Diet–Induced Obesity and Glucose Intolerance
By Jonas Benzler, Goutham K. Ganjam, Dominik Pretz, Rebecca Oelkrug, Christiane E. Koch, Karen Legler, Sigrid Stöhr, Carsten Culmsee, Lynda M. Williams, and Alexander Tups
Introduction
Metabolic inflammation might play a role in the development of overnutrition-induced metabolic syndromes such as obesity, leptin and insulin resistance, and type 2 diabetes. It has previously been shown that upregulation of the central IκB kinase β (IKKβ)/nuclear factor-κB (NF-κB) pathway can cause development of obesity. In this study, both nutritive and genetic inhibition of the central IKKβ/NF-κB pathway were studied to discover whether the metabolic impairments were lessened. The study used diet-induced obese (DIO) and leptin-deficient mice as the subject for the experiment. Some mice were given the known IKKβ/NF-κB inhibitor, dietary flavonoid butein. Other mice experienced genetic inhibition of the pathway in neurons of the arcuate nucleus (ARC), which is known to be a control center of energy homeostasis.
Background
Previous studies established that DIO “in rodents is associated with up-regulation of pro-inflammatory pathways in the hypothalamus, involving activation of signaling intermediates such as…IKKβ/NF-κB” (30). Further, it has been suggested that high-fat diets (HFD) induces metabolic inflammation in the hypothalamus, an important portion of the brain.
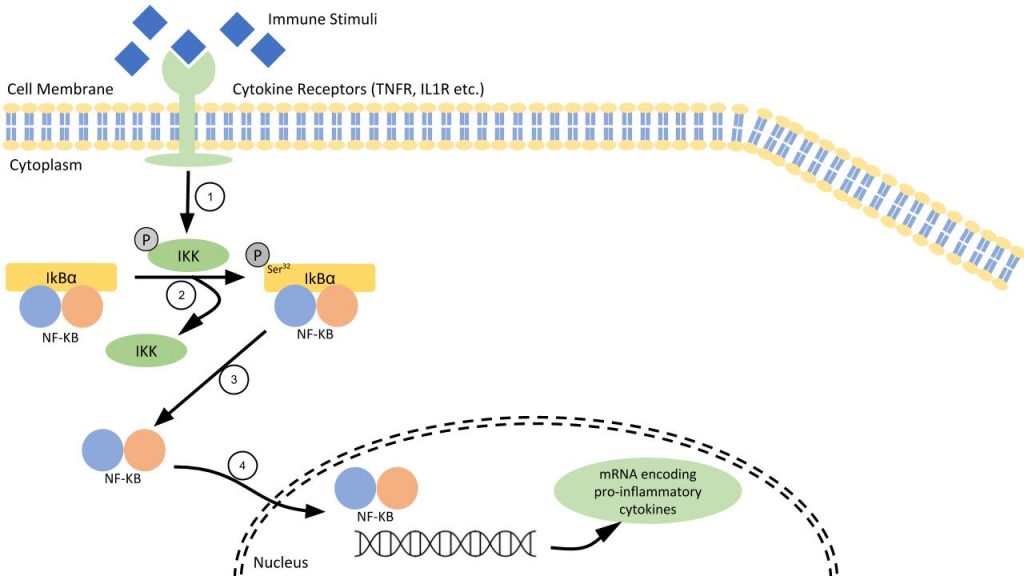
Methods
Experiments were performed on male mice, aged 7 weeks at the beginning of the experiment. The animals had access to a standard chow diet, a low-fat diet, or an HFD.
Stereotaxic Injections: Mice were given intracerebral injections of drugs (butein) through a cannula that was surgically implanted in the left lateral ventricle of the brain.
Results
Flavonoid Butein Inhibition
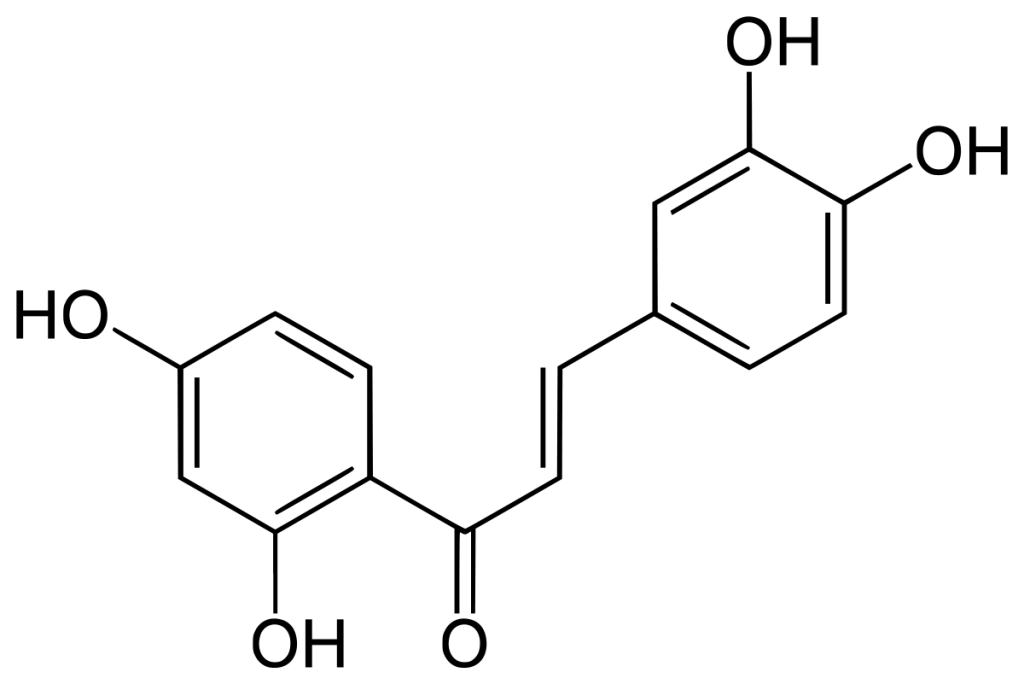
Adapted from Amslinger et al. (31).
Butein directly interacts with a cysteine residue in the activation loop of IKKβ without affecting the phosphorylation status of the molecule. The number of phosphor-IκBα (Ser32) immunoreactive cells was measured in order to see how much inhibition of NF-κB signaling occurred. It was found that, in leptin-deficient mice, acute butein treatment decreased the number of phosphor-IκBα (Ser32), indicating an inhibition of the pathway. A higher dosage (8 mg/kg) of Butein significantly improved glucose tolerance in the mice. Chronic, as opposed to acute, butein treatment was also found to improve blood glucose levels and glucose tolerance.
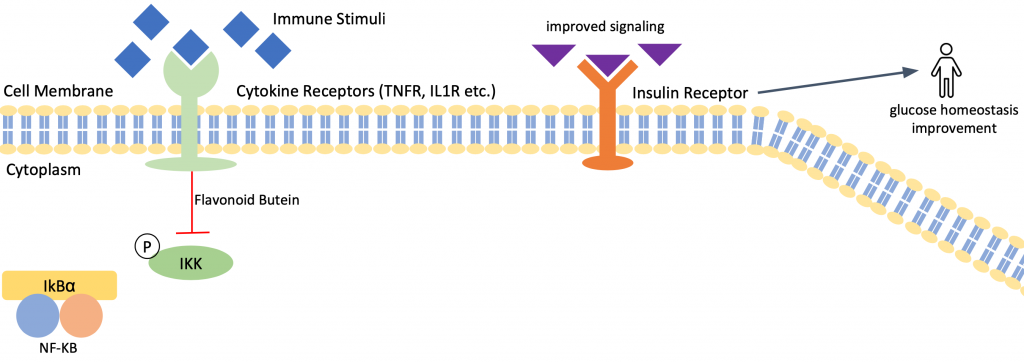
Another group of mice, with DIO, were found to have improved glucose tolerance, as well.
In order to test whether butein acted through mediation of hypothalamic insulin signaling, the activation of the insulin signaling pathway was tested. Butein increased the number of cells positive for phosphor-AKT (Ser473), which is a marker for phosphatidylinositol 3-kinase (PI3K) activation. PI3K, which is in the ARC, is part of the insulin signaling pathway. Inhibition of PI3K prior to butein injection proved that the central PI3K pathway is required for butein to display its antidiabetic properties. Butein’s ability to improve glucose tolerance was fully blocked after the inhibitors were injected.
Overexpression of IκBα
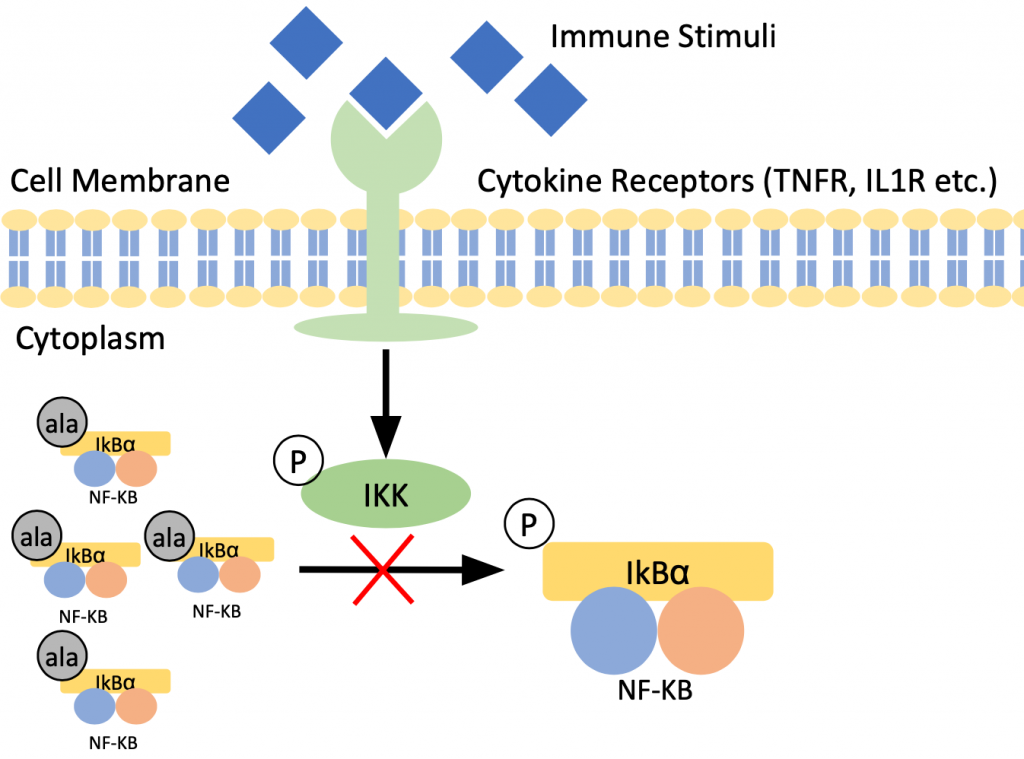
An adeno-associated virus serotype 2 (AAV2) construct allows for overexpression of IκBα mutant in the brain. The inhibitory serine phosphorylation sites of IκBα-mt are replaced by alanine which hinders nuclear translocation of NF- κB.
Overexpression of IκBα-mt was found to reduce DIO. An increase in body weight was induced by an HFD for two groups of mice. The weight gain for the mice that had the IκBα-mt was lower than the control group.
Overexpression of IκBα-mt also improved glucose tolerance in comparison with the controls.
Lastly, overexpression of IκBα-mt altered metabolic rates. The average metabolic rate, measured as oxygen consumption, was increased in mice with IκBα-mt. Additionally, the resting metabolic rate was elevated.
To ensure that overexpression of IκBα-mt altered leptin sensitivity, the expression of a leptin signaling inhibitor was investigated. The action of the inhibitor was decreased with IκBα-mt mice.
Discussion
Prior research on the linkage between obesity and insulin resistance and low-grade inflammation directed the focuses for this paper.
As presumed, flavonoid butein was found to be a glucose-lowering agent. Acute and chronic butein treatment caused a reduction in basal blood glucose levels and improved glucose tolerance. The glucose-lowering effect of butein is mediated via central signaling events within the ARC where butein stabilized the protein IκBα. The PI3K pathway in the ARC is thought to be required for butein action in the brain.
The anti-inflammatory properties of butein are mediated via inhibition of the IKKβ/NF-κB pathway. A gene-therapeutic intervention of IκBα-mt overexpression proved that inhibition of the IKKβ/NF-κB pathway likewise yields anti-inflammatory effects. Overexpression of IκBα-mt minorly reduced HFD-induced weight gain, improved glucose tolerance, and led to reduction in basal blood glucose levels. In summary, overexpression of IκBα-mt inhibited the IKKβ/NF-κB pathway which led to the reduction of weight gain because the pathway encourages inflammation associated with weight gain. When the pathway is inhibited, the inflammation and weight gain is reduced.
The study was not able to prove that viral treatment could reduce total food intake profiles of the mice. The study did show, however, that viral treatment increased energy expenditure due to an elevation in basal energy requirement.
The presence of leptin, a glucose-lowering hormone that sensitizes insulin action, had similar results to butein on glucose levels, suggesting that diet-induced hypothalamic inflammation may be caused by leptin insensitivity. By restoring leptin signaling, the overexpression of IκBα-mt may have the power to enhance insulin sensitivity, and in turn, minimize inflammation.
Summary
Nutritive application of flavonoid butein and gene-therapeutic intervention of IκBα-mt overexpression both inhibited the central IKKβ/NF-κB pathway. Inhibition of the pathway reduced hypothalamic inflammation. The data collected in this study could provide tools to combat HFD-induced metabolic disorders that much of our population faces today.
Recent Comments