Nuclear Factor kappa-B Pathway
Nuclear Factor kappa-B (NF-kB) is a transcription factor that is responsible for triggering the immune response. The NF-kB family is comprised of two classes of proteins. Class 1 consists of NF-kB1 and NF-kB2, and Class 2 consists of RelA, RelB and c-Rel. The difference between the two classes of NF-kB lies in the C-terminus of the structure. Class 1’s C-terminus contains an ankyrin repeat which induces a transrepression activity (binds to silencers, thus inhibiting the expression of the gene). Class 2’s C-terminus has a transactivation function (increases the expression of a gene).
There are two methods of activation of NF-kB, the canonical pathway and the non-canonical pathway. Inflammation relating to obesity revolves around the canonical pathway and thus will be the subject of interest here.
The canonical pathway of NF-kB is activated by a diverse range of external and internal stimuli. However, more importantly to our investigation, pro-inflammatory cytokines such as TNF and IL-1 also activate NF-kB (4).
In its inactive form, NF-kB proteins are sequestered in the cytoplasm by inhibitory proteins such as IkB, more specifically IkBα. Precursors of NF-kB1 and NF-kB2, p105 and p100, also serve a similar function to IkBα as their C-terminal structure is similar to that of IkBα (5).
The binding of the diverse stimuli to its receptors on the cell membrane, such as TNF and IL-1 to TNF receptor (TNFR) and IL-1 receptor (IL1R) respectively, will recruit adaptor proteins which leads to downstream signaling and activation of IkB kinase (IKK – it gets phosphorylated). The IKK complex is made up of two catalytic subunits, IKK1 (IKKα) and IKK2 (IKKβ) and one regulatory subunit NEMO (NF-kB essential modulator, IKKγ). The activation of IKK will then phosphorylate IkBα at the two N-terminal serine which release the NF-kB proteins that were initially sequestered by IkBα (5). The phosphorylated IkBα will undergo proteasome degradation to synthesize new proteins through ubiquitin-dependent IkBα degredation.
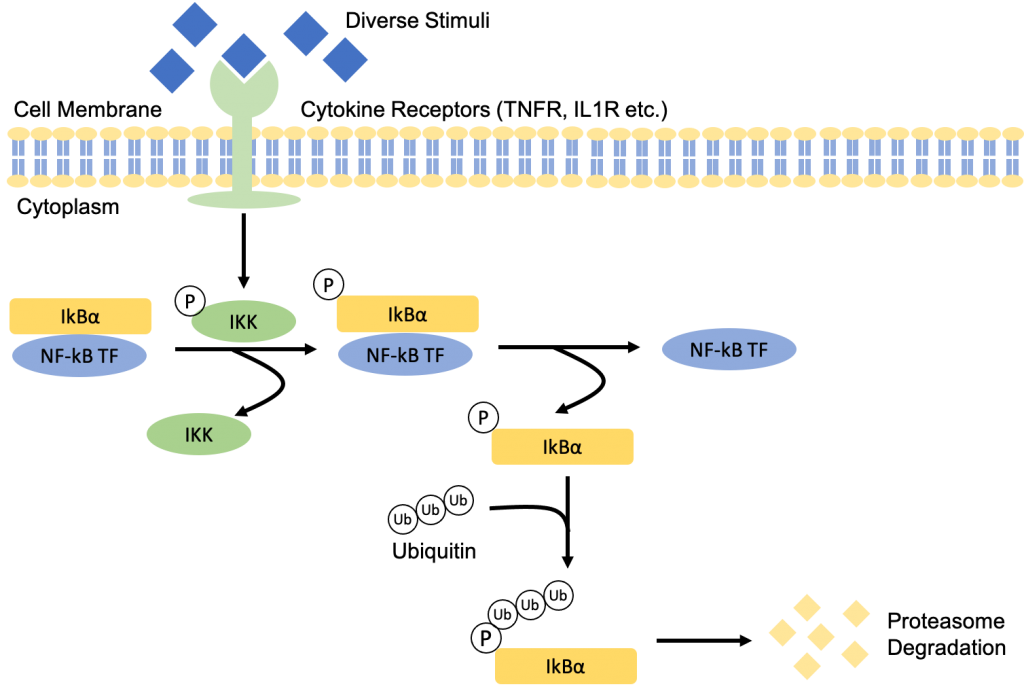
Now that the NF-kB proteins are free, they are able to translocate into the nucleus where they will bind to the promoter sequence and induce transcription expression of various pro-inflammatory cytokine genes (including chemokines) (5).
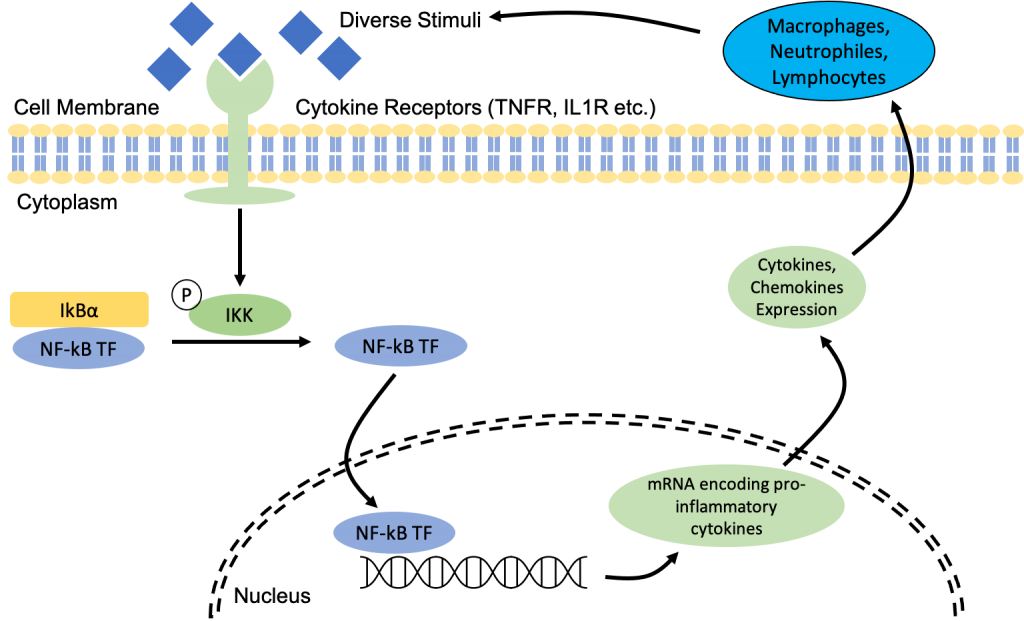
We have previously established that pro-inflammatory cytokines recruit immune cells, and since adipocytes release pro-inflammatory cytokines, there is a relatively high concentration of immune cells within the adipose tissue (as seen here). Immune cells are capable of further activating NF-kB by triggering the release of pro-inflammatory cytokines and recruiting more immune cells through their expression of pattern recognition receptors (PRRs) which are receptors that detect microbial pathogens via pathogen-associated molecular patterns (PAMPs), and fragments of a host cell that is damaged or dead via damage-associated molecular patterns (DAMPs) (5). Even though this involves a completely different pathway, it relates back to the apoptosis of adipocytes mentioned previously and how the constant presence of immune cells such as macrophages, neutrophils and lymphocytes further contribute to the upward cycle of inflammation.
Therefore, cytokines essentially trigger the activation of NF-kB which then results in the production of even more pro-inflammatory cytokines. This continuous production of cytokines results in an upward cycle of inflammation and this is essentially what constitutes as low-grade chronic inflammation.
The activation of IKK also contributes to insulin resistance. When insulin binds to its receptor it recruits a phosphate and phosphorylates insulin receptor substrate proteins 1 and 2 (IRS1/2). These two proteins are tightly regulated by phosphorylation of positive tyrosine and inhibitory serine (22).
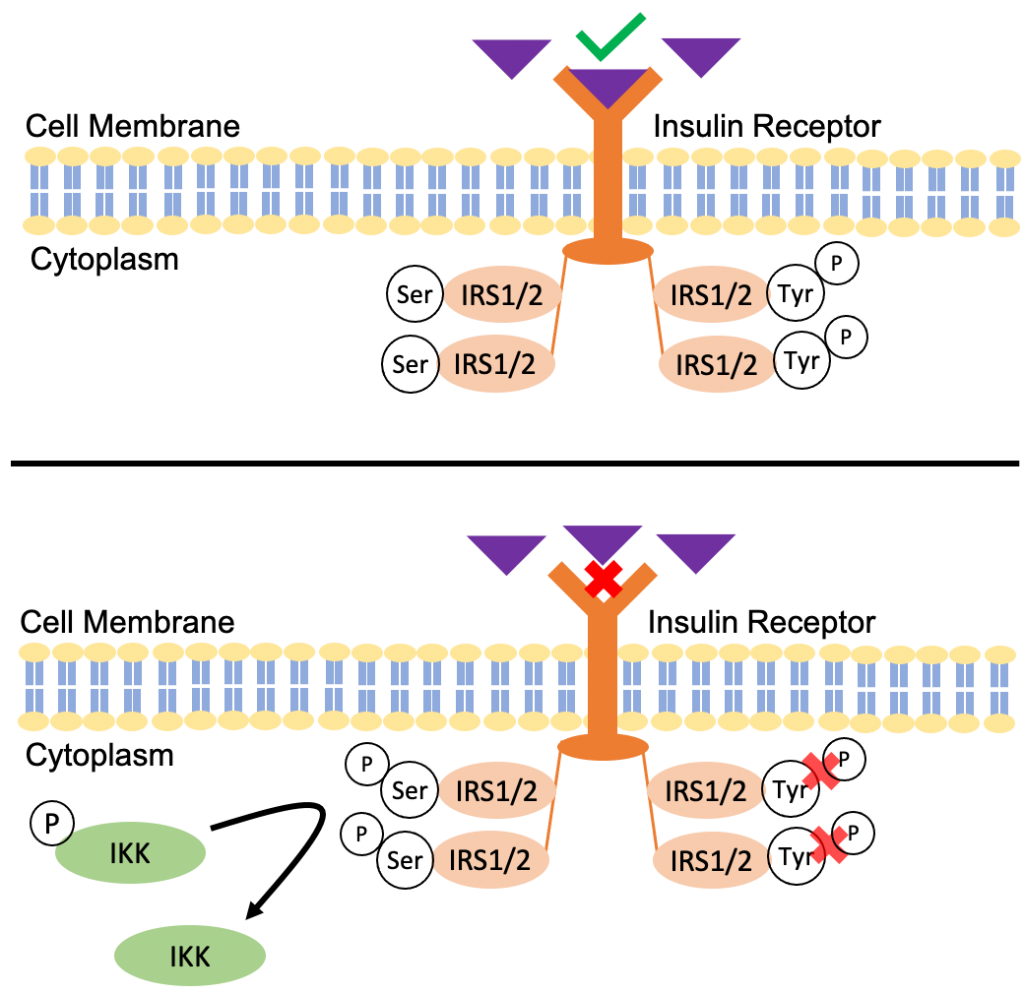
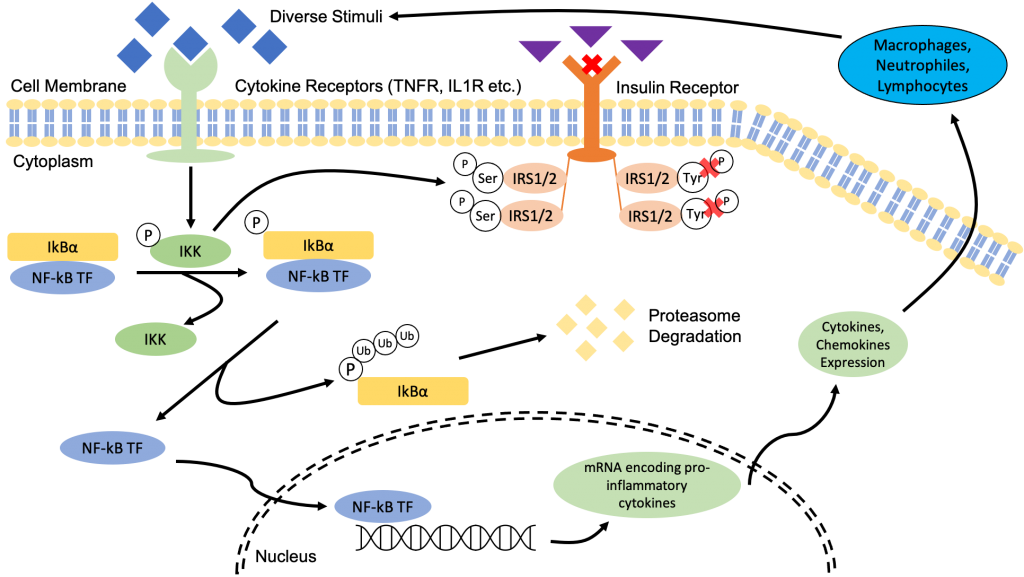
The regular mechanism when insulin binds to its receptor is that it results in an autophosphorylation of tyrosine on the IRS1/2 structure, which triggers the insulin signalling pathway. However, the activation of IKK will phosphorylate the inhibitory serine sites of IRS1/2. As a result, there is a conformational change that prevents the autophosphorylation of tyrosine leading to a decreased sensitivity to insulin signalling, and ultimately insulin resistance (22).
Insulin resistance further contributes to the upward cycle of inflammation as it promotes the intake of food leading to overeating and all the subsequent consequences mentioned in the previous pages. It also increase the secretion of insulin in the bloodstream to make up for the desensitization of insulin. This induces the differentiation of preadipocytes into mature adipocytes prompting hyperplasia (as previously seen here).
Essentially, low-grade chronic inflammation is one huge upward cycle in which the products and several processes of and within the cycle feeds directly back into the start of the cycle.
Recent Comments